sc-OTGM: Single-Cell Perturbation Modeling by Solving Optimal Mass Transport on the Manifold of Gaussian Mixtures

0
🚀
Sign in to get full access
Overview
- Advances in large language models (LLMs) have inspired the development of single-cell foundation models.
- These models have shown success in cell type clustering, phenotype classification, and gene perturbation response prediction.
- However, it's unclear if simpler models could achieve comparable or better results, especially with limited single-cell data.
- Single-cell sequencing data often suffers from technical artifacts, dropout events, and batch effects, which are further complicated by noisy cell state labels in a weakly supervised setting.
Plain English Explanation
Researchers have created new AI models inspired by the recent breakthroughs in large language models. These "single-cell foundation models" can analyze data from individual cells and perform tasks like grouping cells into types, classifying cell properties, and predicting how cells respond to genetic changes.
While these models work well, the researchers wondered if a simpler, more compact model could achieve similar or even better results, especially when dealing with the limited and messy data typical of single-cell experiments. Single-cell data often has technical problems like artifacts and missing information, and the labels used to train the models can be unreliable. These challenges make it difficult to get accurate results.
To address these issues, the researchers developed a new model called sc-OTGM that is much smaller and more efficient than the foundation models, using less than 500,000 parameters. sc-OTGM uses a probabilistic approach to identify distinct cell populations and analyze how genes are regulated in different cell states. It can also generate synthetic single-cell data to supplement real experiments.
Technical Explanation
The researchers present sc-OTGM, a streamlined model with less than 500K parameters, making it approximately 100x more compact than existing single-cell foundation models. sc-OTGM is an unsupervised model that leverages the inductive bias that single-cell RNA sequencing (scRNA-seq) data can be generated from a combination of finite multivariate Gaussian distributions.
The core function of sc-OTGM is to create a probabilistic latent space by utilizing a Gaussian Mixture Model (GMM) as its prior distribution. This allows the model to distinguish between distinct cell populations by learning their respective marginal probability density functions (PDFs). sc-OTGM employs a Hit-and-Run Markov chain sampler to determine the optimal transport (OT) plan across these PDFs within the GMM framework.
The researchers evaluated sc-OTGM on a CRISPR-mediated perturbation dataset called CROP-seq, which consists of 57 single-gene perturbations. Their results demonstrate that sc-OTGM is effective in cell state classification, aids in the analysis of differential gene expression, and ranks genes for target identification through a recommender system. Additionally, sc-OTGM can predict the effects of single-gene perturbations on downstream gene regulation and generate synthetic scRNA-seq data conditioned on specific cell states.
Critical Analysis
While sc-OTGM offers a more efficient alternative to existing single-cell foundation models, the paper acknowledges that the quantity and quality of single-cell data often fall short of the standards seen in textual data used for training large language models. This limitation poses a challenge, as single-cell sequencing data can be affected by technical artifacts, dropout events, and batch effects, which are further complicated by noisy cell state labels in a weakly supervised setting.
Additionally, the paper does not explore the model's performance on more complex tasks, such as multi-source domain adaptation or quantum state generation, which may be necessary to fully assess its capabilities in the single-cell domain. Further research is needed to understand the model's limitations and potential areas for improvement.
Conclusion
The development of sc-OTGM represents a promising step towards more efficient and effective single-cell analysis models. By leveraging a probabilistic approach and a compact architecture, sc-OTGM offers an alternative to the larger foundation models, potentially enabling better performance on limited and noisy single-cell data. This work highlights the continued efforts to address the unique challenges of single-cell sequencing and explores new avenues for advancing the field of computational biology.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
🚀
0
sc-OTGM: Single-Cell Perturbation Modeling by Solving Optimal Mass Transport on the Manifold of Gaussian Mixtures
Andac Demir, Elizaveta Solovyeva, James Boylan, Mei Xiao, Fabrizio Serluca, Sebastian Hoersch, Jeremy Jenkins, Murthy Devarakonda, Bulent Kiziltan
Influenced by breakthroughs in LLMs, single-cell foundation models are emerging. While these models show successful performance in cell type clustering, phenotype classification, and gene perturbation response prediction, it remains to be seen if a simpler model could achieve comparable or better results, especially with limited data. This is important, as the quantity and quality of single-cell data typically fall short of the standards in textual data used for training LLMs. Single-cell sequencing often suffers from technical artifacts, dropout events, and batch effects. These challenges are compounded in a weakly supervised setting, where the labels of cell states can be noisy, further complicating the analysis. To tackle these challenges, we present sc-OTGM, streamlined with less than 500K parameters, making it approximately 100x more compact than the foundation models, offering an efficient alternative. sc-OTGM is an unsupervised model grounded in the inductive bias that the scRNAseq data can be generated from a combination of the finite multivariate Gaussian distributions. The core function of sc-OTGM is to create a probabilistic latent space utilizing a GMM as its prior distribution and distinguish between distinct cell populations by learning their respective marginal PDFs. It uses a Hit-and-Run Markov chain sampler to determine the OT plan across these PDFs within the GMM framework. We evaluated our model against a CRISPR-mediated perturbation dataset, called CROP-seq, consisting of 57 one-gene perturbations. Our results demonstrate that sc-OTGM is effective in cell state classification, aids in the analysis of differential gene expression, and ranks genes for target identification through a recommender system. It also predicts the effects of single-gene perturbations on downstream gene regulation and generates synthetic scRNA-seq data conditioned on specific cell states.
Read more5/8/2024
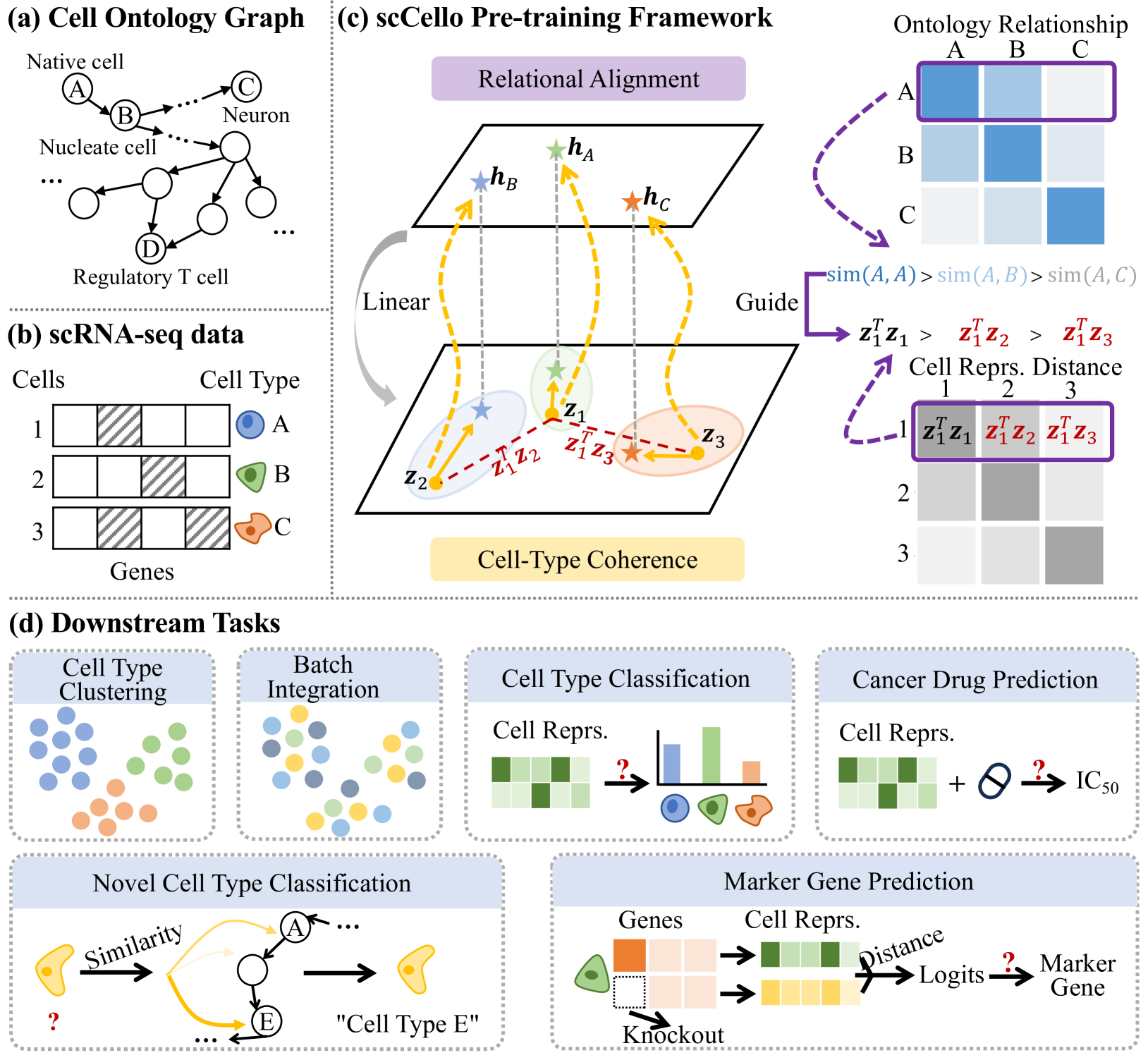
0
Cell-ontology guided transcriptome foundation model
Xinyu Yuan, Zhihao Zhan, Zuobai Zhang, Manqi Zhou, Jianan Zhao, Boyu Han, Yue Li, Jian Tang
Transcriptome foundation models TFMs hold great promises of deciphering the transcriptomic language that dictate diverse cell functions by self-supervised learning on large-scale single-cell gene expression data, and ultimately unraveling the complex mechanisms of human diseases. However, current TFMs treat cells as independent samples and ignore the taxonomic relationships between cell types, which are available in cell ontology graphs. We argue that effectively leveraging this ontology information during the TFM pre-training can improve learning biologically meaningful gene co-expression patterns while preserving TFM as a general purpose foundation model for downstream zero-shot and fine-tuning tasks. To this end, we present textbf{s}ingle textbf{c}ell, textbf{Cell}-textbf{o}ntology guided TFM scCello. We introduce cell-type coherence loss and ontology alignment loss, which are minimized along with the masked gene expression prediction loss during the pre-training. The novel loss component guide scCello to learn the cell-type-specific representation and the structural relation between cell types from the cell ontology graph, respectively. We pre-trained scCello on 22 million cells from CellxGene database leveraging their cell-type labels mapped to the cell ontology graph from Open Biological and Biomedical Ontology Foundry. Our TFM demonstrates competitive generalization and transferability performance over the existing TFMs on biologically important tasks including identifying novel cell types of unseen cells, prediction of cell-type-specific marker genes, and cancer drug responses.
Read more8/23/2024

0
scGHSOM: Hierarchical clustering and visualization of single-cell and CRISPR data using growing hierarchical SOM
Shang-Jung Wen, Jia-Ming Chang, Fang Yu
High-dimensional single-cell data poses significant challenges in identifying underlying biological patterns due to the complexity and heterogeneity of cellular states. We propose a comprehensive gene-cell dependency visualization via unsupervised clustering, Growing Hierarchical Self-Organizing Map (GHSOM), specifically designed for analyzing high-dimensional single-cell data like single-cell sequencing and CRISPR screens. GHSOM is applied to cluster samples in a hierarchical structure such that the self-growth structure of clusters satisfies the required variations between and within. We propose a novel Significant Attributes Identification Algorithm to identify features that distinguish clusters. This algorithm pinpoints attributes with minimal variation within a cluster but substantial variation between clusters. These key attributes can then be used for targeted data retrieval and downstream analysis. Furthermore, we present two innovative visualization tools: Cluster Feature Map and Cluster Distribution Map. The Cluster Feature Map highlights the distribution of specific features across the hierarchical structure of GHSOM clusters. This allows for rapid visual assessment of cluster uniqueness based on chosen features. The Cluster Distribution Map depicts leaf clusters as circles on the GHSOM grid, with circle size reflecting cluster data size and color customizable to visualize features like cell type or other attributes. We apply our analysis to three single-cell datasets and one CRISPR dataset (cell-gene database) and evaluate clustering methods with internal and external CH and ARI scores. GHSOM performs well, being the best performer in internal evaluation (CH=4.2). In external evaluation, GHSOM has the third-best performance of all methods.
Read more7/25/2024
🤿
0
Deep asymmetric mixture model for unsupervised cell segmentation
Yang Nan, Guang Yang
Automated cell segmentation has become increasingly crucial for disease diagnosis and drug discovery, as manual delineation is excessively laborious and subjective. To address this issue with limited manual annotation, researchers have developed semi/unsupervised segmentation approaches. Among these approaches, the Deep Gaussian mixture model plays a vital role due to its capacity to facilitate complex data distributions. However, these models assume that the data follows symmetric normal distributions, which is inapplicable for data that is asymmetrically distributed. These models also obstacles weak generalization capacity and are sensitive to outliers. To address these issues, this paper presents a novel asymmetric mixture model for unsupervised cell segmentation. This asymmetric mixture model is built by aggregating certain multivariate Gaussian mixture models with log-likelihood and self-supervised-based optimization functions. The proposed asymmetric mixture model outperforms (nearly 2-30% gain in dice coefficient, p<0.05) the existing state-of-the-art unsupervised models on cell segmentation including the segment anything.
Read more6/5/2024