Cell-ontology guided transcriptome foundation model

0
Sign in to get full access
Overview
- This paper proposes a cell-ontology guided transcriptome foundation model.
- The model leverages cell ontology information to improve the performance of transformer-based language models on single-cell transcriptomic tasks.
- The authors demonstrate the model's effectiveness on multiple downstream applications, including cell type classification, gene signature prediction, and perturbation modeling.
Plain English Explanation
The researchers have developed a new machine learning model that aims to better understand the genetic information contained in individual cells. This model uses a special type of knowledge about different cell types, called a "cell ontology," to help it learn more effectively from data on gene expression in single cells.
By incorporating this cell ontology information, the model is able to perform better on various tasks related to analyzing single-cell gene expression data. For example, it can more accurately classify the types of cells present in a sample, predict which genes are active in different cell types, and model how cells respond to various biological perturbations.
The key idea is that the cell ontology provides useful context and structure that helps the model make better use of the single-cell gene expression data it is trained on. This leads to improved performance compared to more standard machine learning approaches that don't leverage this additional information.
Overall, this work represents an important advance in developing more powerful computational tools for extracting insights from the vast amount of single-cell genomic data being generated by modern biological research.
Technical Explanation
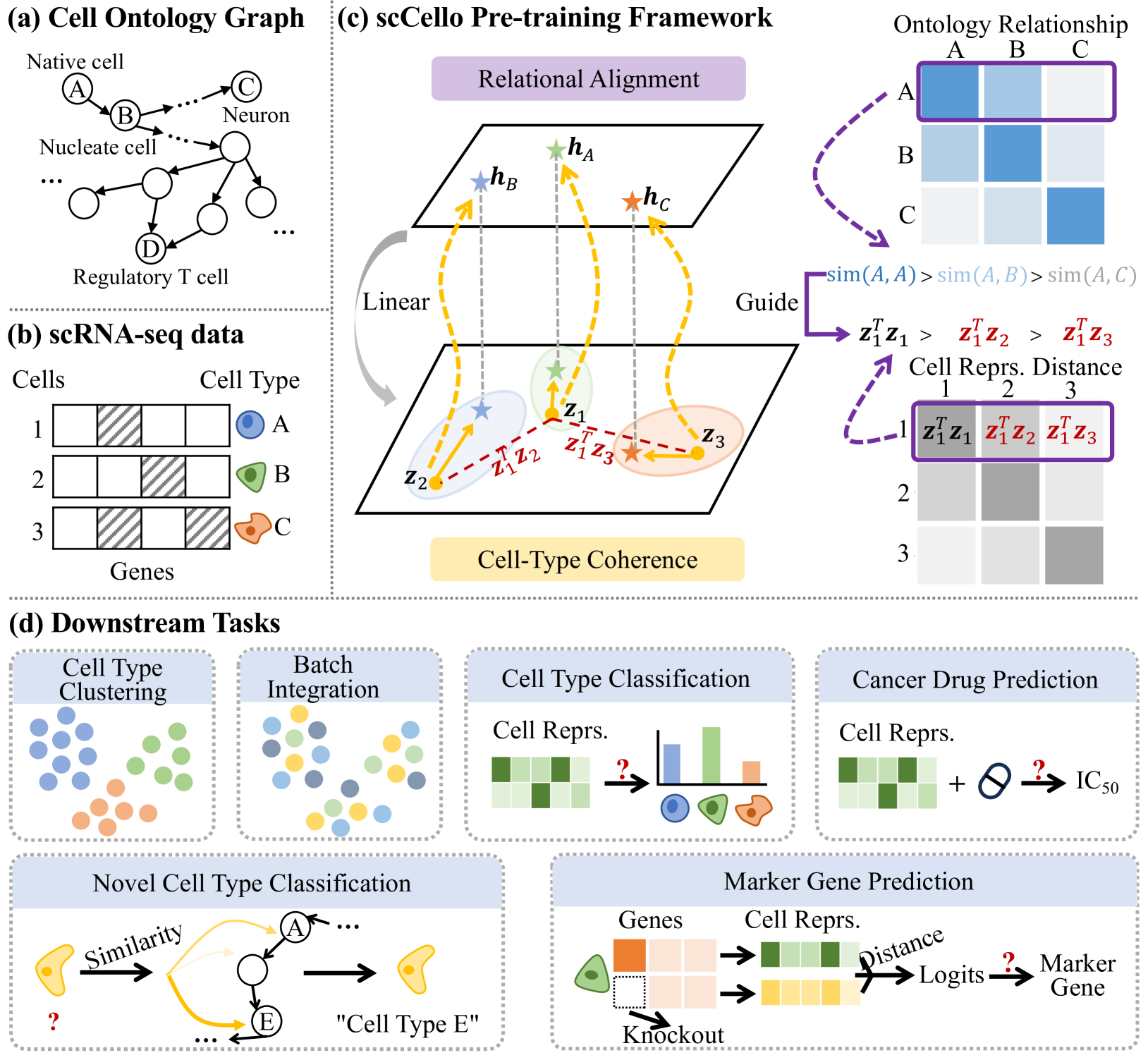
The paper introduces a cell-ontology guided transcriptome foundation model. This model builds on top of transformer-based language models, but integrates knowledge from the Cell Ontology, a comprehensive taxonomy of cell types.
The authors pre-train the model on a large corpus of single-cell transcriptomic data, using the cell ontology to guide the representation learning process. This allows the model to better capture the underlying biological structures and relationships between different cell types and their gene expression profiles.
The pre-trained model is then fine-tuned on various downstream tasks, such as:
The authors demonstrate that this cell-ontology guided approach leads to significant performance improvements compared to models that do not leverage this additional structural information.
Critical Analysis
The paper presents a well-designed and thorough evaluation of the proposed model, with experiments on multiple challenging single-cell transcriptomic tasks. The authors acknowledge several limitations, such as the reliance on the curated Cell Ontology and the need for further research on the interpretability of the model's learned representations.
One potential concern is the generalizability of the approach, as the performance gains may be sensitive to the specific ontology used or the characteristics of the training data. It would be valuable to explore the model's robustness to variations in cell ontologies or the inclusion of additional modalities beyond just gene expression.
Additionally, while the authors highlight the model's potential for understanding biological mechanisms, more work may be needed to fully realize this capability and connect the learned representations to meaningful biological insights.
Conclusion
This paper introduces an innovative approach to leveraging structured domain knowledge, in the form of a cell ontology, to enhance the performance of transformer-based models on single-cell transcriptomic tasks. The results demonstrate the value of incorporating such prior information and suggest that this cell-ontology guided foundation model could be a valuable tool for advancing research in single-cell biology and genomics.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Cell-ontology guided transcriptome foundation model
Xinyu Yuan, Zhihao Zhan, Zuobai Zhang, Manqi Zhou, Jianan Zhao, Boyu Han, Yue Li, Jian Tang
Transcriptome foundation models TFMs hold great promises of deciphering the transcriptomic language that dictate diverse cell functions by self-supervised learning on large-scale single-cell gene expression data, and ultimately unraveling the complex mechanisms of human diseases. However, current TFMs treat cells as independent samples and ignore the taxonomic relationships between cell types, which are available in cell ontology graphs. We argue that effectively leveraging this ontology information during the TFM pre-training can improve learning biologically meaningful gene co-expression patterns while preserving TFM as a general purpose foundation model for downstream zero-shot and fine-tuning tasks. To this end, we present textbf{s}ingle textbf{c}ell, textbf{Cell}-textbf{o}ntology guided TFM scCello. We introduce cell-type coherence loss and ontology alignment loss, which are minimized along with the masked gene expression prediction loss during the pre-training. The novel loss component guide scCello to learn the cell-type-specific representation and the structural relation between cell types from the cell ontology graph, respectively. We pre-trained scCello on 22 million cells from CellxGene database leveraging their cell-type labels mapped to the cell ontology graph from Open Biological and Biomedical Ontology Foundry. Our TFM demonstrates competitive generalization and transferability performance over the existing TFMs on biologically important tasks including identifying novel cell types of unseen cells, prediction of cell-type-specific marker genes, and cancer drug responses.
Read more8/23/2024

0
LangCell: Language-Cell Pre-training for Cell Identity Understanding
Suyuan Zhao, Jiahuan Zhang, Yushuai Wu, Yizhen Luo, Zaiqing Nie
Cell identity encompasses various semantic aspects of a cell, including cell type, pathway information, disease information, and more, which are essential for biologists to gain insights into its biological characteristics. Understanding cell identity from the transcriptomic data, such as annotating cell types, has become an important task in bioinformatics. As these semantic aspects are determined by human experts, it is impossible for AI models to effectively carry out cell identity understanding tasks without the supervision signals provided by single-cell and label pairs. The single-cell pre-trained language models (PLMs) currently used for this task are trained only on a single modality, transcriptomics data, lack an understanding of cell identity knowledge. As a result, they have to be fine-tuned for downstream tasks and struggle when lacking labeled data with the desired semantic labels. To address this issue, we propose an innovative solution by constructing a unified representation of single-cell data and natural language during the pre-training phase, allowing the model to directly incorporate insights related to cell identity. More specifically, we introduce $textbf{LangCell}$, the first $textbf{Lang}$uage-$textbf{Cell}$ pre-training framework. LangCell utilizes texts enriched with cell identity information to gain a profound comprehension of cross-modal knowledge. Results from experiments conducted on different benchmarks show that LangCell is the only single-cell PLM that can work effectively in zero-shot cell identity understanding scenarios, and also significantly outperforms existing models in few-shot and fine-tuning cell identity understanding scenarios.
Read more6/12/2024
0
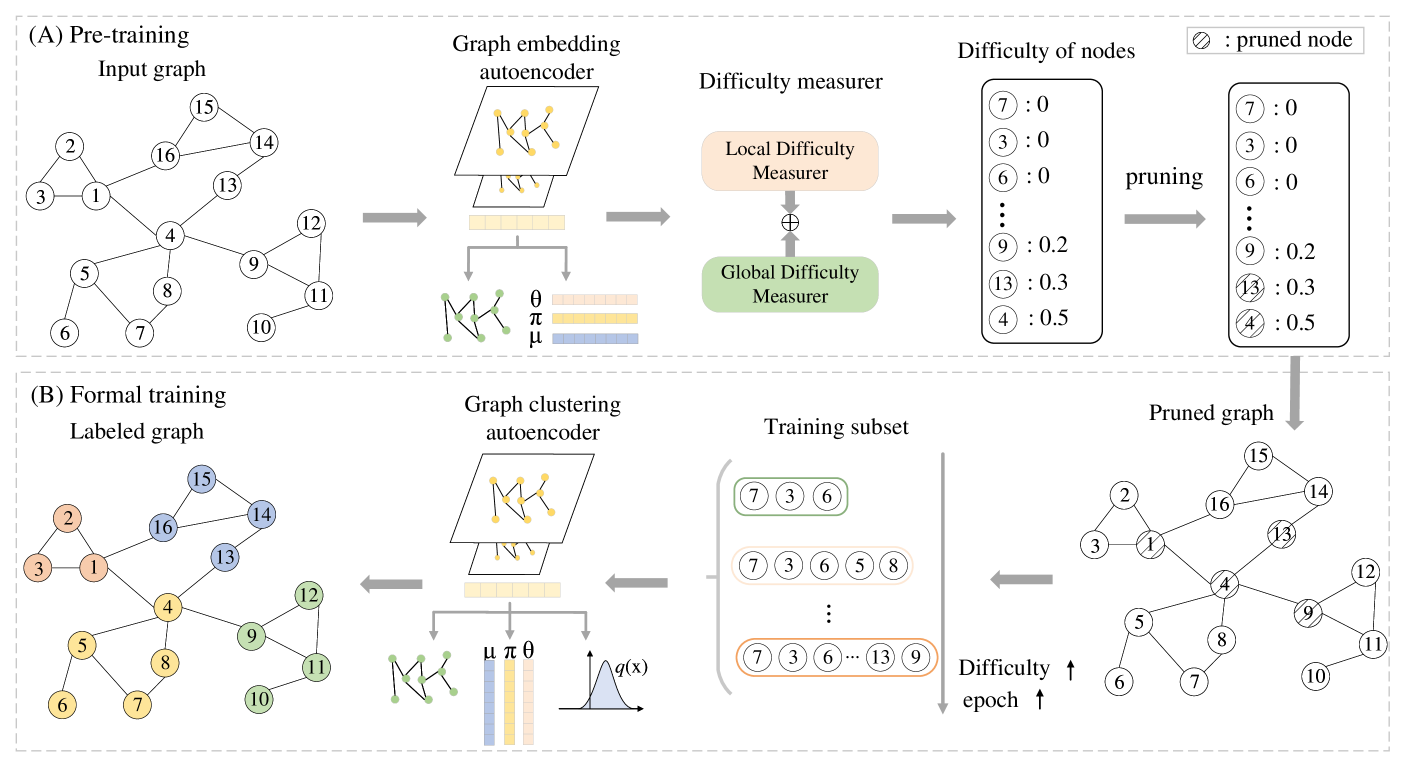
Single-cell Curriculum Learning-based Deep Graph Embedding Clustering
Huifa Li, Jie Fu, Xinpeng Ling, Zhiyu Sun, Kuncan Wang, Zhili Chen
The swift advancement of single-cell RNA sequencing (scRNA-seq) technologies enables the investigation of cellular-level tissue heterogeneity. Cell annotation significantly contributes to the extensive downstream analysis of scRNA-seq data. However, The analysis of scRNA-seq for biological inference presents challenges owing to its intricate and indeterminate data distribution, characterized by a substantial volume and a high frequency of dropout events. Furthermore, the quality of training samples varies greatly, and the performance of the popular scRNA-seq data clustering solution GNN could be harmed by two types of low-quality training nodes: 1) nodes on the boundary; 2) nodes that contribute little additional information to the graph. To address these problems, we propose a single-cell curriculum learning-based deep graph embedding clustering (scCLG). We first propose a Chebyshev graph convolutional autoencoder with multi-decoder (ChebAE) that combines three optimization objectives corresponding to three decoders, including topology reconstruction loss of cell graphs, zero-inflated negative binomial (ZINB) loss, and clustering loss, to learn cell-cell topology representation. Meanwhile, we employ a selective training strategy to train GNN based on the features and entropy of nodes and prune the difficult nodes based on the difficulty scores to keep the high-quality graph. Empirical results on a variety of gene expression datasets show that our model outperforms state-of-the-art methods.
Read more8/21/2024

0
Tissue Concepts: supervised foundation models in computational pathology
Till Nicke, Jan Raphael Schaefer, Henning Hoefener, Friedrich Feuerhake, Dorit Merhof, Fabian Kiessling, Johannes Lotz
Due to the increasing workload of pathologists, the need for automation to support diagnostic tasks and quantitative biomarker evaluation is becoming more and more apparent. Foundation models have the potential to improve generalizability within and across centers and serve as starting points for data efficient development of specialized yet robust AI models. However, the training foundation models themselves is usually very expensive in terms of data, computation, and time. This paper proposes a supervised training method that drastically reduces these expenses. The proposed method is based on multi-task learning to train a joint encoder, by combining 16 different classification, segmentation, and detection tasks on a total of 912,000 patches. Since the encoder is capable of capturing the properties of the samples, we term it the Tissue Concepts encoder. To evaluate the performance and generalizability of the Tissue Concepts encoder across centers, classification of whole slide images from four of the most prevalent solid cancers - breast, colon, lung, and prostate - was used. The experiments show that the Tissue Concepts model achieve comparable performance to models trained with self-supervision, while requiring only 6% of the amount of training patches. Furthermore, the Tissue Concepts encoder outperforms an ImageNet pre-trained encoder on both in-domain and out-of-domain data.
Read more9/6/2024